In the Nov. 4, 2010 issue of Nature, there is an interesting Comment, titled "Transparency showcases strength of peer review", by Bernd Pulverer, head of scientific publications at the European Molecular Biology Organization and chief editor of The EMBO Journal. In this article, Pulverer "reflects on his experience at The EMBO Journal of publishing referees’ reports, authors’ responses and editors’ comments alongside papers."
The peer-review process of scientific articles has traditionally been a "black box": (anonymous) reviewers' reports, editors' comments, and authors' responses – extremely valuable information in shaping the final form of published papers – are all hidden from public view. In the Internet era, technology (e.g., online space) is no longer an issue. Now The EMBO Journal has led the way, and "the experience has been overwhelmingly positive." Hopefully, other leading journals (e.g., Nature and Science) would follow the example. Afterall, making the peer-review process transparent is an excellent mean to increase the accountability of science and scientific publications.
Overall, this article is well-written, succinct and logical, and it touches an important topic in scientific publication. Over the past few days, I have read several "Review Process Files" accompanying papers I am interested in, e.g., "Recognition of the amber UAG stop codon by release factor RF1", and found them highly revealing.
Saturday, November 6, 2010
Saturday, October 30, 2010
Publication of scientific programming code
Recently on the Nature website, I read with great interest a news article, titled "Publish your computer code: it is good enough", by Nick Barnes, a professional software engineer:
Freely provided working code — whatever its quality — improves programming and enables others to engage with your researchClearly the author knows the "trade secret" in scientific programming. He lists several common reasons why scientists are reluctant to share their source code, and then provides his responses:
- The code is low quality — "software in all trades is written to be good enough for the job intended". All software has bugs. Sharing code would help improve the code itself and advance the research field.
- Not a common practice — this is going to change or is already changing.
- Demand for support — "Nobody is entitled to demand technical support for freely provided code."
- Intellectual property issue — The most value part "lies in your expertise", code not backed by skilled experts is called abandonware. (I cannot agree more with this point.)
- Polishing code takes time/effort — not need to, just supply, as supplementary materials in a website, the original code used in your publication.
Saturday, October 23, 2010
Chi (χ) torsion angle characterizes base/sugar relative orientation
Except for pseudouridine, a nucleoside in DNA/RNA contains an N-glycosidic bond that connects the base to the sugar. The chi (χ) torsion angle, which characterizes the relative base/sugar orientation, is defined by O4'-C1'-N1-C2 for pyrimidines (C, T and U), and O4'-C1'-N9-C4 for purines (A and G).
Normally (as in A- and B-form DNA/RNA duplex), χ falls into the ranges of +90° to +180°; –90° to –180° (or 180° to 270°), corresponding to the anti conformation (Figure below, top). Occasionally, χ has values in the range of –90° to +90°, referring to the syn conformation (Figure below, bottom). Note that in left-handed Z-DNA with CG repeating sequence, the purine G is in syn conformation whilst the pyrimidine C is anti.

Presumably, the χ-related anti/syn conformation is a very basic and simple concept. In essence, though, the N-glycosidic bond and the corresponding χ torsion angle illustrate that the base and sugar are two separate entities, i.e. there is an internal degree of freedom between them. In this respect, it is worth noting that the Leontis-Westhod sugar edge for base-pair classification corresponds to the anti form only. When a base is flipped over into the syn conformation, the "sugar edge", defined in connection with the minor (shallow) groove side of a nitrogenous bases, simply does not exist.
Base-flipping (anti/syn conformation switch) is one of the factors associated with the two possible relative orientations in a base pair, characterized explicitly in 3DNA as of type M+N or M–N since the 2003 NAR paper (Figure 2, linked below). I reemphasized this distinction in our 2010 GpU dinucleotide platform paper (in particular, see supplementary Figure S2). Unfortunately, this subtle (but crucial, in my opinion) point has never been taken seriously (or at all) by the RNA community, even with 3DNA's wide adoption. However, as people know 3DNA deeper/better and take RNA base-pair classification more rigorously, I have no doubt they will begin to appreciate the simplicity of this explicit distinction and the resultant full quantification of each and every possible base pair using standard geometric parameters.

On a related issue, current versions of 3DNA (v1.5 and v2.0) output only the χ torsion angle without providing the anti/syn classification. This defect, and many others, will hopefully be rectified in future releases of 3DNA.
Normally (as in A- and B-form DNA/RNA duplex), χ falls into the ranges of +90° to +180°; –90° to –180° (or 180° to 270°), corresponding to the anti conformation (Figure below, top). Occasionally, χ has values in the range of –90° to +90°, referring to the syn conformation (Figure below, bottom). Note that in left-handed Z-DNA with CG repeating sequence, the purine G is in syn conformation whilst the pyrimidine C is anti.

Presumably, the χ-related anti/syn conformation is a very basic and simple concept. In essence, though, the N-glycosidic bond and the corresponding χ torsion angle illustrate that the base and sugar are two separate entities, i.e. there is an internal degree of freedom between them. In this respect, it is worth noting that the Leontis-Westhod sugar edge for base-pair classification corresponds to the anti form only. When a base is flipped over into the syn conformation, the "sugar edge", defined in connection with the minor (shallow) groove side of a nitrogenous bases, simply does not exist.
Base-flipping (anti/syn conformation switch) is one of the factors associated with the two possible relative orientations in a base pair, characterized explicitly in 3DNA as of type M+N or M–N since the 2003 NAR paper (Figure 2, linked below). I reemphasized this distinction in our 2010 GpU dinucleotide platform paper (in particular, see supplementary Figure S2). Unfortunately, this subtle (but crucial, in my opinion) point has never been taken seriously (or at all) by the RNA community, even with 3DNA's wide adoption. However, as people know 3DNA deeper/better and take RNA base-pair classification more rigorously, I have no doubt they will begin to appreciate the simplicity of this explicit distinction and the resultant full quantification of each and every possible base pair using standard geometric parameters.

On a related issue, current versions of 3DNA (v1.5 and v2.0) output only the χ torsion angle without providing the anti/syn classification. This defect, and many others, will hopefully be rectified in future releases of 3DNA.
Friday, October 15, 2010
Improving the design of existing code by refactoring
Another software engineering book I read recently is "Refactoring: Improving the Design of Existing Code" by Martin Fowler. According to the author,
The book is practical in nature; it not just explains the principles but provides a detailed account of over 70 commonly used refactorings. As vividly explained by the author in the first paragraph of Chapter 1, "Refactoring, a First Example", "it is with examples that I [the author] can see what is going on." This approach fits my style perfectly: to really understand a topic, I always find a worked example far more effective than general principles. While the examples in the book are illustrated in Java, the basic ideas can be applied well to other object-oriented or even procedural languages (such as C).
Over the years since I left Dr. Olson's laboratory at Rutgers, I have been maintaining and continuously refining 3DNA. I have taken each user's question as an opportunity to fix bugs and improve its design, thus making the code more robust and efficient. The majority of my efforts, as I now realize, is "refactoring" existing 3DNA code to make it easier to maintain and extend. Reading through this book gives me the chance to put my practices into a broader context. I will surely take advantage of some refactoring examples from the book for further refinements of 3DNA.
Refactoring is the process of changing a software system in such a way that it does not alter the external behavior of the code yet improve its internal structure. It is a disciplined way to clean up code that minimize the chances of introducing bugs. In essence when you refractor you are improving the design of the code after it has been written.
The book is practical in nature; it not just explains the principles but provides a detailed account of over 70 commonly used refactorings. As vividly explained by the author in the first paragraph of Chapter 1, "Refactoring, a First Example", "it is with examples that I [the author] can see what is going on." This approach fits my style perfectly: to really understand a topic, I always find a worked example far more effective than general principles. While the examples in the book are illustrated in Java, the basic ideas can be applied well to other object-oriented or even procedural languages (such as C).
Over the years since I left Dr. Olson's laboratory at Rutgers, I have been maintaining and continuously refining 3DNA. I have taken each user's question as an opportunity to fix bugs and improve its design, thus making the code more robust and efficient. The majority of my efforts, as I now realize, is "refactoring" existing 3DNA code to make it easier to maintain and extend. Reading through this book gives me the chance to put my practices into a broader context. I will surely take advantage of some refactoring examples from the book for further refinements of 3DNA.
Wednesday, October 13, 2010
NSMB editorial: "Go figure"
In the October 2010 issue of Nature Structural & Molecular Biology (NSMB, Vol. 17, No. 10) there is another interesting one-page editorial, titled "Go figure", which provides tips on how to make a scientific figure that may worth 1000 words:
As pointed out by the author, "These are just a few guidelines and suggestions for handling figures." Overall, "simplicity rules in scientific figures, as in life." I guess no one would argue with such general advices. However, it would be even more helpful to illustrate such points with concrete examples (I know that seems to be beyond the scope of a one-page editorial).
A picture may be worth a thousand words, but ensuring that those words make sense is important, especially in the context of a scientific figure. Here are some tips for making your figures count.A recap of the tips is given below; by and large, they all follow conventional wisdom:
- General considerations: Each figure should make just one point and be self-explanatory.
- See guidelines. "At all stages, the figures should be clear and legible."
- How many figures? The figures should complement the Results section, and be included only necessary.
- How many panels? Better only one; multiple panels "should be logically connected."
- What’s in a label? Keep it succinct, but make the figure self-explanatory.
- Getting colorful. Use color wisely and constantly.
- A legendary figure. The figure legend should concise and informative.
- A model paper. Better have a figure (at the end) of the final model that conveys "the big picture". Honestly, I do not quite get this point.
Thursday, September 30, 2010
Further details of the DNA story revealed by Crick's lost correspondence
In the September 30 issue of Nature (Vol. 467, pp519-524), there is an interesting account of "The lost correspondence of Francis Crick" by Gann & Witkowski. The newly found letters, mostly between Crick and Wilkins, unveil further background information on the exciting DNA story. As the authors put it, "Strained relationships and vivid personalities leap off the pages."
I read Watson's "The Double Helix" book a while ago, and overall I am quite familiar with the DNA story. Still, I found this account fascinating: it provides a "CAST LIST" in "The search for the structure of DNA" with photos (p521); and it succinctly summarizes the relationships among the key players. In science, no other story shows more dramatically the collaborative and competitive nature among scientists working on similar projects.

Franklin’s X-ray diffraction photograph 51 of B-form DNA (see figure above, from Wikipedia), with its unambiguous evidence that DNA was helical, proved crucial for Watson and Crick to determine the structure of DNA. Indeed, the Watson-Crick DNA model corresponds to the B-form DNA, with its base-pairs in the middle, parallel to each other and perpendicular to the linear helical axis.
From this Nature account, however, I noticed for the first time a subtle detail: when B-form DNA photograph 51 was shown to Watson by Wilkins in early 1953, Franklin also already had the A-form DNA diffraction pattern. According to the authors,
When Crick had the opportunity to look the A-form DNA diffraction picture, on 5 June 1953, he wrote (to Wilkins):
I am reading "Blink: The Power of Thinking Without Thinking", a book by Malcolm Gladwell. The above case serves as a vivid example.
I read Watson's "The Double Helix" book a while ago, and overall I am quite familiar with the DNA story. Still, I found this account fascinating: it provides a "CAST LIST" in "The search for the structure of DNA" with photos (p521); and it succinctly summarizes the relationships among the key players. In science, no other story shows more dramatically the collaborative and competitive nature among scientists working on similar projects.

Franklin’s X-ray diffraction photograph 51 of B-form DNA (see figure above, from Wikipedia), with its unambiguous evidence that DNA was helical, proved crucial for Watson and Crick to determine the structure of DNA. Indeed, the Watson-Crick DNA model corresponds to the B-form DNA, with its base-pairs in the middle, parallel to each other and perpendicular to the linear helical axis.
From this Nature account, however, I noticed for the first time a subtle detail: when B-form DNA photograph 51 was shown to Watson by Wilkins in early 1953, Franklin also already had the A-form DNA diffraction pattern. According to the authors,
It was the A-structure diffraction pattern that had led Franklin away from believing that DNA, in that form at least, was helical, despite her already having produced the most persuasive helical pictures of the B structure — including photograph 51. The crystalline DNA gave better quality diffraction data, more suited to her painstaking, quantita- tive approach, and so she focused on the A form during 1952. It was at this time that she and Gosling made a handwritten, black edged funeral card announcing the death of “DNA Helix(crystalline)”.
When Crick had the opportunity to look the A-form DNA diffraction picture, on 5 June 1953, he wrote (to Wilkins):
This is the first time I have had an opportunity for a detailed study of the picture of Structure A, and I must say I am glad I didn’t see it earlier, as it would have worried me considerably.
I am reading "Blink: The Power of Thinking Without Thinking", a book by Malcolm Gladwell. The above case serves as a vivid example.
Sunday, September 19, 2010
New NDB entries have IDs start with the prefix NA?
Recently, while reading the article titled "Designing Triple Helical Fragments: The Crystal Structure of the Undecamer d(TGGCCTTAAGG) Mimicking T.AT Base Triplets" by Van Hecke (Crystal Growth & Design), I noticed the following:
Over the years, NDB has established itself prominently as "a repository of three-dimensional structural information about nucleic acids". Traditionally, an NDB id has some associated "meanings", with noticeably exceptions, as discussed one year ago in my blog post "PDB id vs NDB id". Thus, it is not surprising that NDB decided to make changes in naming new ids. However, I cannot find any announcement on the id change in the NDB website; a Google search on "NDB id" did not uncover anything new either. Luckily, NDB provides search by release date ("Released Since"). After a few tries, I traced that the new id policy began to be implemented from around March 2010.
The new NDB id convention appears to be NA (presumably standing for for Nucleic Acid) followed by 4 digits. As more entries available, it is not that hard to imagine that the number of digits must be expanded to 5, 6, or even more. Naturally, PDB id, with a fixed 4-character length (up to now), is (far) more consistent than the NDB id is.
The atomic coordinates and structure factors have been deposited in the Protein Data Bank41 and Nucleic Acid Database42 (PDB and NDB entry codes 3L1Q and NA0392, respectively).The NDB id NA0392 reminded me of an email communication I had with Dr. Olson early this year when she told me of the id change of new NDB entries. Occupied with other issues, I did not pay much attention to this point until recently.
Over the years, NDB has established itself prominently as "a repository of three-dimensional structural information about nucleic acids". Traditionally, an NDB id has some associated "meanings", with noticeably exceptions, as discussed one year ago in my blog post "PDB id vs NDB id". Thus, it is not surprising that NDB decided to make changes in naming new ids. However, I cannot find any announcement on the id change in the NDB website; a Google search on "NDB id" did not uncover anything new either. Luckily, NDB provides search by release date ("Released Since"). After a few tries, I traced that the new id policy began to be implemented from around March 2010.
The new NDB id convention appears to be NA (presumably standing for for Nucleic Acid) followed by 4 digits. As more entries available, it is not that hard to imagine that the number of digits must be expanded to 5, 6, or even more. Naturally, PDB id, with a fixed 4-character length (up to now), is (far) more consistent than the NDB id is.
Sunday, September 12, 2010
"Code Complete", a practical handbook of software construction
Over the summer, I read quite a few books on C programming, and more generally on software construction. Among them, the book titled "Code Complete" (2nd edition) by Steve McConnell is the most comprehensive: with over 900 pages, it certainly serves as "a practical handbook of software construction".
I have been writting scientific software applications for over twenty years, using a variety of programming languages. Gradually, writing code per se is taking less time; nowadays by far the most time-consuming parts have increasingly become (1) to understand a scientific topic thoroughly in order to implement it in new code, and (2) to maintain/refine/adapt existing codebase to meet the needs of its user community (e.g., 3DNA). It is for the benefit of the later that I've found the "Code Complete" book very helpful.
As an example, following the book's recommendations (1) "Use boolean variables to simplify complicated tests" (pp301-2) and (2) "Simplify complicated tests with boolean function calls" (p359), I've easily improved the readability of some parts of my code.
Overall, the book is well-written and full of practical advices; I enjoyed reading it.
I have been writting scientific software applications for over twenty years, using a variety of programming languages. Gradually, writing code per se is taking less time; nowadays by far the most time-consuming parts have increasingly become (1) to understand a scientific topic thoroughly in order to implement it in new code, and (2) to maintain/refine/adapt existing codebase to meet the needs of its user community (e.g., 3DNA). It is for the benefit of the later that I've found the "Code Complete" book very helpful.
As an example, following the book's recommendations (1) "Use boolean variables to simplify complicated tests" (pp301-2) and (2) "Simplify complicated tests with boolean function calls" (p359), I've easily improved the readability of some parts of my code.
Overall, the book is well-written and full of practical advices; I enjoyed reading it.
Sunday, September 5, 2010
Identification of C-H...N/O H-honds using 3DNA
Recently, I came across an interesting article by Kiliszek et al., titled "Atomic resolution structure of CAG RNA repeats: structural insights and implications for the trinucleotide repeat expansion diseases". In addition to its biological implications, this paper uses 3DNA to deal with non-canonical-base-pair-containing helical structures in a sensible way:
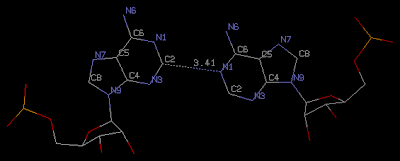
The find_pair program in the currently distributed versions of 3DNA (v2.0 and before), however, does not identify this A-A pair (thus the corresponding NDB list of "Base Pair Step Parameters" is incomplete) for the following two reasons:
The helical parameters were calculated using 3DNA (29). Sequence-independent measures were used, based on vectors connecting the C1' atoms of the paired residues, to avoid computational artefacts arising from non-canonical base pairing.Another significant point, which is the focus of this post, is the observation that "All the adenosines are in the anti-conformation and the only interaction within each A-A pair is a single C2-H2...N1 hydrogen bond." (Figure below) Given the 0.95 Å ultrahigh resolution of structure 3nj6/na0608, it is likely that this type of A-A is real.

The find_pair program in the currently distributed versions of 3DNA (v2.0 and before), however, does not identify this A-A pair (thus the corresponding NDB list of "Base Pair Step Parameters" is incomplete) for the following two reasons:
- No H-bond exists between N/O base atoms – currently a requirement for a base-pair.
- By default, only N/O atoms are used in defining H-bonds (see tag hb_atoms in file "misc_3dna.par"). Nevertheless, by adding C as a possible atom in forming H-bond, and manually editing find_pair generated input file to analyze, 3DNA structural parameters can be calculated as usual.
1 95 # 1 | ...1>A:...1_:[..G]G-----C[..C]:..10_:A<...2
2 94 # 2 | ...1>A:...2_:[..G]G-----C[..C]:...9_:A<...2
3 93 # 3 | ...1>A:...3_:[..C]C-----G[..G]:...8_:A<...2
4 92 # 4 | ...1>A:...4_:[..A]A-**--A[..A]:...7_:A<...2
5 91 # 5 | ...1>A:...5_:[..G]G-----C[..C]:...6_:A<...2
6 90 # 6 | ...1>A:...6_:[..C]C-----G[..G]:...5_:A<...2
7 89 # 7 | ...1>A:...7_:[..A]A-**--A[..A]:...4_:A<...2
8 88 # 8 | ...1>A:...8_:[..G]G-----C[..C]:...3_:A<...2
9 87 # 9 | ...1>A:...9_:[..C]C-----G[..G]:...2_:A<...2
10 86 # 10 | ...1>A:..10_:[..C]C-----G[..G]:...1_:A<...2
##### Criteria: 4.00 0.00 15.00 2.50 65.00 4.50 7.50 [ O N C]
##### 2 non-Watson-Crick base-pairs, and 1 helix (0 isolated bps)
##### Helix #1 (10): 1 - 10
Subscribe to:
Comments (Atom)
